
不容錯(cuò)過:超全面的材料成分分析方法及典型應(yīng)用舉例!
2018-12-14 18:30:20
作者:本網(wǎng)整理 來源:有色技術(shù)平臺(tái)
分享至:


1 前言
材料科學(xué)的主要內(nèi)容就是研究材料的組成與結(jié)構(gòu)、形貌與缺陷等對(duì)材料性能的影響,而這些研究與材料分析方法緊密相關(guān)[1]。材料分析可分為三大方面:材料結(jié)構(gòu)的測(cè)定、材料形貌觀察和材料成分分析[2]。材料成分分析主要是指通過各種檢測(cè)手段對(duì)樣品的成分進(jìn)行定性定量的分析。常用的分析方法主要有化學(xué)分析法、光譜法、、X射線能量色散譜法(EDX)和電子能譜法,下面對(duì)這幾種方法一一介紹。
2 化學(xué)分析法
利用物質(zhì)的化學(xué)反應(yīng)為基礎(chǔ)的分析,稱為化學(xué)分析。每種物質(zhì)都有其獨(dú)特的化學(xué)特性,因此我們可以利用物質(zhì)間的化學(xué)反應(yīng)并將其以一種適當(dāng)?shù)姆绞竭M(jìn)行表征用以指示反應(yīng)的進(jìn)程,從而得到材料中某些組分的含量[3]。
2.1 滴定法
滴定法又可稱為容量分析法。如圖1所示,在該法中,滴定標(biāo)準(zhǔn)溶液通過滴定管逐漸加到待測(cè)物中,直至反應(yīng)完全,然后測(cè)量消耗的標(biāo)準(zhǔn)溶液的體積就可以計(jì)算出待測(cè)物質(zhì)的含量[4]。滴定法要求滴定反應(yīng)應(yīng)有足夠大的反應(yīng)平衡常數(shù)而且反應(yīng)速率足夠快。即是說,滴加的標(biāo)準(zhǔn)溶液應(yīng)當(dāng)與待測(cè)物完全而又迅速的發(fā)生反應(yīng),直至待測(cè)物被消耗完畢。常見的滴定法可分為三類:酸堿滴定法、氧化還原滴定法和沉淀滴定法。滴定法不需要特別的儀器,成本低,而且準(zhǔn)確性高。
PEI J[5]提出了一種測(cè)定鈦粉中鐵含量的滴定方法。該法是使用二苯胺磺酸鈉作為指示劑,然后使用標(biāo)準(zhǔn)K2Cr2O7溶液進(jìn)行滴定從而測(cè)定鐵含量。該方法的原理是:Fe2+在溶液中呈現(xiàn)為黃色,當(dāng)使用K2Cr2O7進(jìn)行滴定時(shí)其就被氧化為紫紅色的Fe3+,據(jù)此可以判斷出滴定終點(diǎn)。與采用分光光度法,ICP-AES法相比,重鉻酸鉀滴定測(cè)定鐵含量最為準(zhǔn)確,但該法操作上稍顯復(fù)雜。
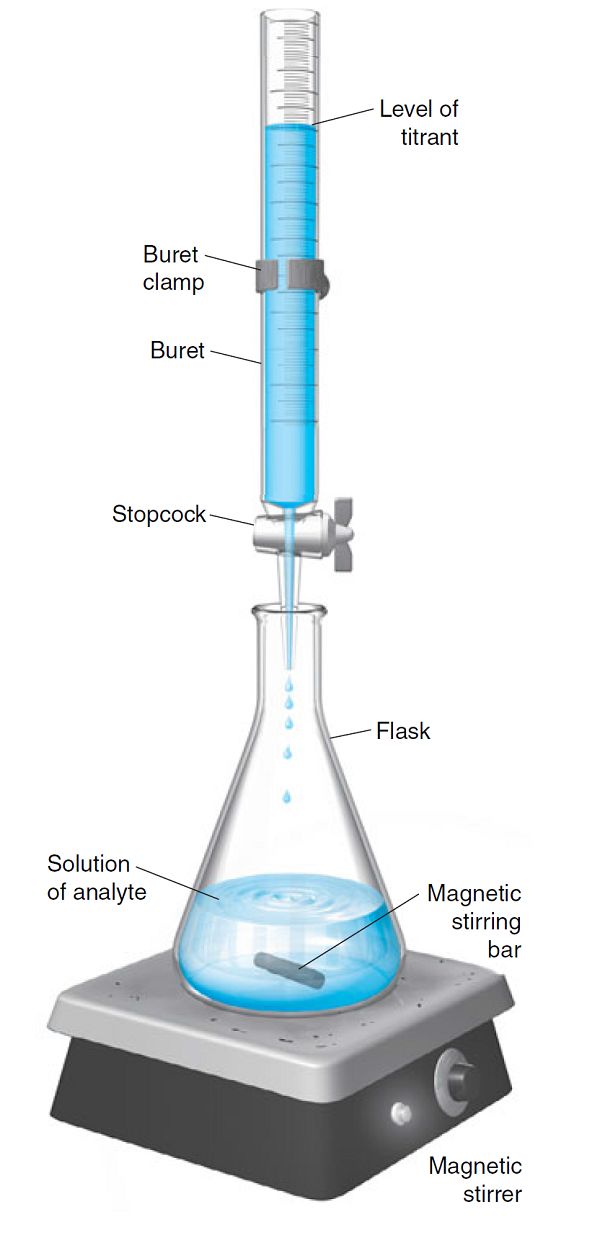
圖1 滴定法實(shí)驗(yàn)裝置示意圖[4]。
2.2 重量法
重量法是使用產(chǎn)物的質(zhì)量來計(jì)算初始物質(zhì)的含量[4]。根據(jù)物質(zhì)的化學(xué)性質(zhì),選擇合適的化學(xué)反應(yīng),將被測(cè)組分轉(zhuǎn)化為一種組成固定的沉淀或氣體形式,通過鈍化、干燥、灼燒或吸收劑的吸收等一系列的處理后,精確稱量,從而求出被測(cè)組分的含量。重量法對(duì)儀器的要求不高,成本比較低,結(jié)果準(zhǔn)確,不過稍顯繁瑣。
陳敏芳等[6]就提出使用喹鉬檸酮啉重量法測(cè)定磷肥中的磷含量。其主要原理是在酸性介質(zhì)中,正磷酸根與喹鉬檸酮沉淀劑反應(yīng)生成黃色磷鉬酸喹啉沉淀,將沉淀過濾洗滌,干燥稱重即可計(jì)算出材料中的磷含量[7]。鋰離子電池正極材料LiFePO4中磷的含量也常常使用該法測(cè)量。
重量法的另一大技術(shù)是熱重分析(TG)。它是指在程序控制溫度下,測(cè)量物質(zhì)質(zhì)量與溫度之間的關(guān)系的技術(shù)。根據(jù)材料的性質(zhì),通過分析熱重曲線,就可以知道被測(cè)物質(zhì)在多少度時(shí)產(chǎn)生變化,并且根據(jù)失重量,可以計(jì)算失去了多少物質(zhì)(如CuSO4·5H2O中的結(jié)晶水,見圖2)。
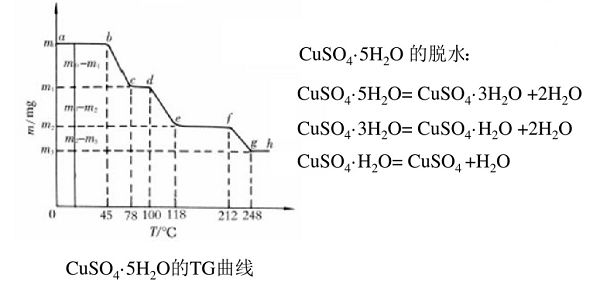
圖2 硫酸銅的失重曲線。
熱重分析在文獻(xiàn)中的應(yīng)用非常之多,其中一個(gè)最廣泛的應(yīng)用是測(cè)定復(fù)合材料的碳含量。Tahir等[8]制備一種TiO2和碳的復(fù)合材料用作鈉離子電池負(fù)極,為了得到TiO2的含量,作者采用了TG對(duì)樣品進(jìn)行了分析,結(jié)果如圖3所示。在0-200 ℃的失重可認(rèn)為是吸附水的脫去引起的,而350-550 ℃重量的急劇下降則是因?yàn)樘既紵兂蒀O2造成的。550 ℃以后樣品的質(zhì)量不再變化,這是因?yàn)門iO2在空氣中是穩(wěn)定的。據(jù)此,可以分析出吸附水,碳和TiO2的含量分別為2%,10%和88%。
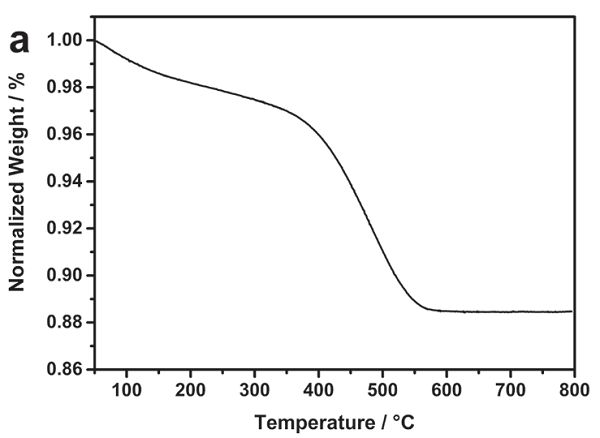
圖3 TiO2-C復(fù)合材料的失重曲線。
2.3 燃燒分析法
燃燒分析就是將樣品在過量氧氣中進(jìn)行燃燒,樣品經(jīng)高溫氧化燃燒生成氮?dú)狻⒌难趸铩⒍趸肌⒍趸蚝退龋⒃谳d氣的推動(dòng)下,進(jìn)入分離檢測(cè)單元。該法通常用于測(cè)量有機(jī)化合物中的C、H、O、N和S[4]。現(xiàn)代有機(jī)元素分析儀(Elemental analysis,簡(jiǎn)稱EA)就是據(jù)此制造的,與傳統(tǒng)的化學(xué)元素分析方法相比,元素分析儀自動(dòng)化程度高,操作簡(jiǎn)便迅速,測(cè)量C、H、N、S時(shí)的精確度均小于0.2%,已逐漸成為元素分析的主要方法之一。需要注意的是,由于吸附水的存在,樣品中H和O的含量通常難以測(cè)準(zhǔn)。此外,由于是采用燃燒的方式進(jìn)行樣品處理,因此,易爆炸的樣品測(cè)試時(shí)也需要特別注意。

圖4 有機(jī)元素分析的原理
LiFePO4是一種具有較大發(fā)展?jié)摿Φ匿囯x子電池正極材料,但是其較低的電子電導(dǎo)率阻礙了其性能的發(fā)揮。為了提高鋰離子電池正極材料LiFePO4的電子電導(dǎo)率,人們常常在其表面包覆碳。然而不同的碳含量對(duì)其電池材料的性能有著較大的影響[9],因此測(cè)量所合成材料的碳含量就顯得很重要。Jing Du等[10]就利用CHS元素分析儀對(duì)其制備的多孔的碳包覆的LiFePO4中的碳含量進(jìn)行了測(cè)定,結(jié)果顯示C含量為6.5 wt%。此外,元素分析也常常用于Li-S電池中含硫材料中硫含量的測(cè)定。
3 原子光譜法
原子光譜是原子吸收或發(fā)出(發(fā)光和熒光)光子的強(qiáng)度關(guān)于光子能量(通常以波長(zhǎng)表示)的圖譜,其可以提供關(guān)于樣品化學(xué)組成的相關(guān)信息[11]。原子光譜分為三大類:原子吸收光譜、原子發(fā)射光譜和原子熒光光譜[12]。由于原子吸收光譜和原子發(fā)射光譜應(yīng)用最為廣泛,故主要介紹此兩種。
3.1 原子發(fā)射光譜(OES/AES)
原子發(fā)射光譜是一種很古老的技術(shù),通常用于元素分析[13, 14]。每種元素的原子及離子激發(fā)后,都會(huì)輻射出一組表征該元素的特征光譜線。其中有一條或數(shù)條輻射的強(qiáng)度最強(qiáng),最容易被檢測(cè)出來,所以也常常稱為最靈敏線。根據(jù)元素靈敏線的出現(xiàn)與否就可以確定樣品中是否存在這些元素,這就是OES定性分析的基本原理。在一定條件下,元素的特征譜線的強(qiáng)度隨著元素在樣品中的含量或濃度的增大而增強(qiáng),利用這一性質(zhì)可以測(cè)定元素的含量,這就是OES定量分析的依據(jù)。
原子發(fā)射光譜儀器主要由光源、樣品室、光學(xué)色散系統(tǒng)和檢測(cè)器等組成[15],現(xiàn)在最常用的是ICP-OES(電感耦合等離子體原子發(fā)射光譜)。ICP-OES是使用電感耦合等離子體做為光源。使用電感耦合等離子體作為光源是因?yàn)槠溆幸恍┆?dú)特的性質(zhì)[16-18],它能產(chǎn)生更高的溫度并減少可能會(huì)發(fā)生的化學(xué)反應(yīng)且其穩(wěn)定性好,這會(huì)使OES線性分析范圍廣,靈敏度高,化學(xué)干擾低,更加適用于光譜定性分析和定量分析。OES廣泛應(yīng)用的主要原因就是其通用性強(qiáng)和具有多元素分析能力。
OES有著很高的精確度,相對(duì)標(biāo)準(zhǔn)偏差(%RSD)只有1%或更低。ICP-OES幾乎可以檢測(cè)元素周期表中的所有元素,檢測(cè)限低至0.1~50 ?g/l,但是對(duì)堿金屬來說其檢測(cè)限卻比較差,因?yàn)榈入x子體的溫度對(duì)這些元素來說太高了,高的溫度也使其存在較嚴(yán)重的譜線干擾問題。ICP-OES可以用來分析任何能制成溶液的樣品,然而其缺點(diǎn)也在于樣品需要制成溶液,這對(duì)于固體樣品來說不僅費(fèi)時(shí)而且繁瑣。一般來說,ICP-OES主要用于能溶解的樣品中無機(jī)多元素的分析[11]。
鄒本東等[19]等就使用該方法測(cè)定了褐煤中鍺和一些主要成灰元素。作者選取了適宜的儀器工作條件、選擇了無干擾的分析譜線和合理地扣除了光譜背景,樣品灰化后用HNO3/HF/HClO4混合酸消解,測(cè)定結(jié)果與國(guó)標(biāo)方法吻合。此外作者還對(duì)同一煤樣共6份,用來計(jì)算相對(duì)標(biāo)準(zhǔn)偏差即為方法的精密度,并按照IUPAC定義得到檢出限,結(jié)果表明測(cè)試相對(duì)標(biāo)準(zhǔn)偏差小于5%,準(zhǔn)確度符合要求。
3.2 原子吸收光譜(AAS)
原子吸收光譜法又稱為原子吸收分光光度法。每種元素都有其特征的光譜線,當(dāng)光源發(fā)射的某一特征波長(zhǎng)的光通過待測(cè)樣品的原子蒸氣時(shí),原子中的外層電子將選擇性地吸收其同種元素所發(fā)射的特征譜線,使光源發(fā)出的入射光減弱,吸光度與被測(cè)樣品中的待測(cè)元素含量成正比。通過測(cè)定吸收的光量,就可以求出樣品中待測(cè)的金屬及類金屬物質(zhì)的含量這就是原子吸收光譜分析法的原理[20]。
如圖5所示,原子吸收光譜儀是由光源、原子化系統(tǒng)、光學(xué)系統(tǒng)、檢測(cè)系統(tǒng)和顯示裝置五大部分組成的,其中原子化系統(tǒng)在整個(gè)裝置中具有至關(guān)重要的作用[21]。
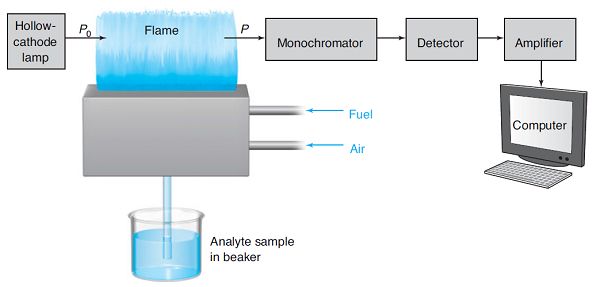
圖5 原子吸收光譜實(shí)驗(yàn)示意圖[4]
原子吸收光譜法,選擇性強(qiáng),因其原子吸收的譜線僅發(fā)生在主線系,且譜線很窄,所以光譜干擾小、選擇性強(qiáng)、測(cè)定快速簡(jiǎn)便、靈敏度高。電熱原子吸收光譜法的檢測(cè)限低至1 ?g/l[11],分析范圍廣,目前可測(cè)定元素多達(dá)73種;既可測(cè)定液態(tài)樣品,又可測(cè)定氣態(tài)或某些固態(tài)樣品。原子吸收光譜法譜線的強(qiáng)度受溫度影響較小,精密度高,常規(guī)低含量測(cè)定時(shí),精密度為1%~3%。
ICP-OES相比,原子吸收光譜法不能對(duì)多元素同時(shí)進(jìn)行分析,但是操作卻更簡(jiǎn)單、成本更低,因此在檢測(cè)含量在?g/l的樣品時(shí)常常使用AAS。AAS對(duì)難溶元素的測(cè)定靈敏度也不十分令人滿意,對(duì)共振譜線處于真空紫外區(qū)的元素,如P、S等還無法測(cè)定。另外,標(biāo)準(zhǔn)工作曲線的線性范圍窄,給實(shí)際工作帶來不便,對(duì)于某些復(fù)雜樣品的分析,還需要進(jìn)一步消除干擾。
考慮到AAS的優(yōu)點(diǎn),張輝等[22]就使用AAS測(cè)定了蔬菜中礦物元素的含量。他使用的是澳大利亞GBC公司Avanta PM型原子吸收分光光度計(jì)。首先作者制定了待測(cè)元素的校準(zhǔn)曲線,然后對(duì)通過酸消解處理的蔬菜樣品進(jìn)行了測(cè)定,通過校準(zhǔn)曲線計(jì)算了相應(yīng)元素的含量。此外作者還測(cè)試了樣品的加標(biāo)回收率。試驗(yàn)表明加標(biāo)回收率在94.0%~106%之間,這也說明方法準(zhǔn)確可行。
3.3 X射線熒光光譜(XRF)
X射線熒光光譜分析(X Ray Fluorescence, XRF)的原理是采用X射線管產(chǎn)生的入射X射線(又叫一次X射線或原級(jí)X射線)激發(fā)被測(cè)樣品,隨后受激發(fā)樣品中的元素就會(huì)發(fā)射出二次X射線(又稱X射線熒光),并且不同的元素所放射出的二次X射線具有特定的能量特性或波長(zhǎng)特性。探測(cè)系統(tǒng)測(cè)量這些放射出來的二次X射線的能量及數(shù)量,就可將其轉(zhuǎn)換成各種元素的種類及含量(如圖6)。利用X射線熒光原理,理論上可以測(cè)量元素周期表中Be以后的每一種元素。在實(shí)際應(yīng)用中,有效的元素測(cè)量范圍為9號(hào)元素 (F)到92號(hào)元素(U)。
XRF分析的特點(diǎn)是適合于各類固體樣品中主、次、痕量多元素同時(shí)檢測(cè),檢出限約在μg/g量級(jí)范圍內(nèi),制樣方法簡(jiǎn)單,現(xiàn)已廣泛應(yīng)用于地質(zhì)、材料、環(huán)境、冶金樣品的常規(guī)分析。XRF是一種無損檢測(cè)技術(shù),可直接應(yīng)用于現(xiàn)場(chǎng)、原位及活體分析。XRF的缺點(diǎn)是檢出限不夠低,不適用于分析輕元素,準(zhǔn)確定量分析依賴標(biāo)樣。XRF的另一個(gè)缺點(diǎn)是需要的樣品較多,粉末一般需要2g以上。
崔梅生等[23]就采用手持 XRF 技術(shù)對(duì)汽車尾氣催化劑、汽車尾氣催化劑粉體、金屬整體式摩托車尾氣催化劑等樣品進(jìn)行貴金屬含量測(cè)試。與常規(guī) ICP-OES 方法相比,手持 XRF 技術(shù)是一種快速、方便、廉價(jià)的貴金屬含量測(cè)試方法,對(duì)于汽車尾氣催化劑粉體,手持 XRF 檢測(cè)與 ICP-OES 認(rèn)證值對(duì)于 Pd 含量相對(duì)標(biāo)準(zhǔn)偏差小于5%。
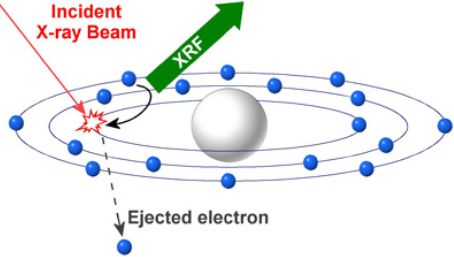
圖6 XRF的基本原理
4 X射線能量色散譜法(EDX)
EDX常與電子顯微鏡配合使用,它是測(cè)量電子與試樣相互作用所產(chǎn)生的特征X射線的波長(zhǎng)與強(qiáng)度,從而對(duì)微小區(qū)域所含元素進(jìn)行定性或定量分析[24]。每一種元素都有一個(gè)特定波長(zhǎng)的特征X射線與之相對(duì)應(yīng),它不隨入射電子的能量而變化,測(cè)量電子激發(fā)試樣所產(chǎn)生的特征X射線波長(zhǎng)的種類,即可確定試樣中所存在元素的種類。元素的含量與該元素產(chǎn)生的特征X射線強(qiáng)度成正比,據(jù)此可以測(cè)定元素的含量。
EDX可分析表面1~5μm的元素成分,探測(cè)靈敏度大約為0.1%(原子摩爾分?jǐn)?shù),與元素種類有關(guān))[25]。EDX可探測(cè)元素范圍大(4Be~92U),對(duì)重元素的分析特別有效。它可以同時(shí)測(cè)量所有的元素,分析效率高[26]。能譜所需探針電流小,對(duì)待分析試樣損傷小,適于生物試樣、快離子導(dǎo)體試樣等。
EDX有三種基本工作方式。一是定點(diǎn)分析,對(duì)樣品表面選定微區(qū)進(jìn)行掃描分析;二是線掃描分析,可以對(duì)樣品表面選定的直線進(jìn)行元素定性定量分析;三是面掃描分析,可以獲得某種元素質(zhì)量分布的掃描圖像。由于EDX通常是SEM或者TEM的附件,因此,采用EDX進(jìn)行元素分析的一大優(yōu)勢(shì)就是可視化操作,非常直觀。
但是能譜儀分辨率較差,峰背比低,譜峰重疊嚴(yán)重[27]。此外,能譜儀工作條件要求嚴(yán)格,需要液氮冷卻[12]。由于電子進(jìn)入的深度的限制,EDX只能用來分析局部表層的元素種類和含量,不適用于定量分析,精度有時(shí)較大。
Jang-Zern Tsai等[28]就使用EDX表征了COOH-P-SPCE(富羧基多孔絲網(wǎng)碳電極)表面修飾前的元素分布和元素含量。結(jié)果如圖7所示,分析表明COOH-P-SPCE表面C和Ca的含量分別為90.38wt%和9.62 wt%,結(jié)合SEM圖像也可以發(fā)現(xiàn)元素在表面分布也比較均勻。
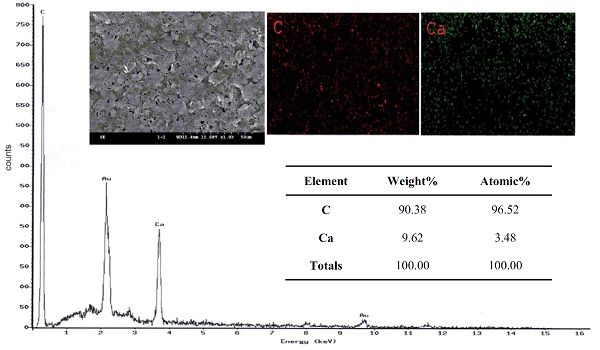
圖7 COOH-P-SPCE的SEM圖像和EDS圖譜[26]
5 電子能譜分析法
電子能譜分析法是采用單色光源或電子束去照射樣品,使樣品中電子受到激發(fā)而發(fā)射出來,然后測(cè)量這些電子的產(chǎn)額(強(qiáng)度)與能量的分布,從而獲得材料信息。電子能譜的采樣深度僅為幾納米,所以其僅是表面成分的反應(yīng)[29]。本文主要介紹應(yīng)用較為廣泛的俄歇電子能譜法和X射線光電子能譜法。
5.1 俄歇電子能譜法(AES)
當(dāng)原子吸收X射線或者被電子轟擊時(shí),如果在其內(nèi)部產(chǎn)生了空穴,原子就有可能發(fā)射俄歇電子(如圖8所示),通過檢測(cè)俄歇電子的能量和強(qiáng)度就可以獲得樣品表面化學(xué)成分和結(jié)構(gòu)的相關(guān)信息[30]。
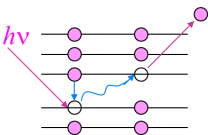
圖8 俄歇電子發(fā)射示意圖
俄歇電子能譜可以用來對(duì)元素進(jìn)行定性分析,主要是利用俄歇電子的特征能量值來確定固體表面的元素組成[31],方法是將測(cè)得的譜圖與標(biāo)準(zhǔn)譜圖進(jìn)行對(duì)比。俄歇電子能譜可以用以定量分析,但因?yàn)橛绊懚硇盘?hào)強(qiáng)弱的因素很多,導(dǎo)致其定量分析比較復(fù)雜,因此俄歇能譜分析精度較低,基本上是半定量的水平,一般情況下相對(duì)精度僅為30%。
AES的優(yōu)點(diǎn)是在距表面0.5~2nm范圍內(nèi)靈敏度高、分析速度快,能探測(cè)周期表上H和He以外的所有元素[1]。由于采樣深度僅為在2nm以內(nèi),所以其更適合于表面元素定性分析,配合離子束剝離技術(shù),AES還有很強(qiáng)的深度分析(深度分析速度和分辨率都比XPS好[29])和界面分析能力。AES擁有者與XPS相同的表面靈敏度而且相對(duì)簡(jiǎn)單,但準(zhǔn)確程度較XPS差[32]。此外,通過顯微AES還可以獲得俄歇電子能譜元素分布圖(SAM),但由于該分析方法耗時(shí)非常長(zhǎng),一般很少使用。
AES也有一些局限性。首先它不能用于分析H和He元素。其次它的定量分析的準(zhǔn)確度不高。另外其對(duì)多數(shù)元素的探測(cè)靈敏度為0.1%~1.0%(原子摩爾分?jǐn)?shù))。此外電子束轟擊損傷和電荷積累問題也限制了其在有機(jī)材料、生物樣品和一些陶瓷材料中的應(yīng)用。俄歇電子能譜對(duì)樣品的要求高,表面必須清潔。
尹燕萍等[33]使用AES配合氬離子束剝離技術(shù)研究了樣品的元素剖面分布。圖9就是是用于制作CCD器件的PtSi/Si樣品的AES深度分析圖。由圖中的深度剖面分布看,Si上的PtSi層約4 nm厚,而且界面處有較嚴(yán)重的氧吸附。
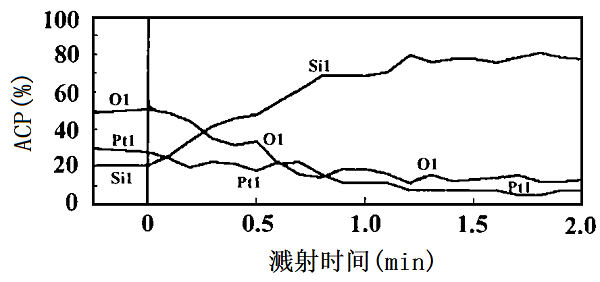
圖9 PtSi/Si的AES組分深度分布。
5.2X射線光電子能譜法(XPS)
XPS是由瑞典Uppsala大學(xué)的Kaiser Siegbahn及其同事基于光電效應(yīng)的原理研制發(fā)展的的一種表面化學(xué)分析技術(shù)[34-37]。XPS使用軟X射線作為激發(fā)光源,激發(fā)出物質(zhì)表面原子的內(nèi)層電子(如圖10),通過對(duì)這些電子進(jìn)行能量分析而獲得材料信息。

更多關(guān)于材料方面、材料腐蝕控制、材料科普等方面的國(guó)內(nèi)外最新動(dòng)態(tài),我們網(wǎng)站會(huì)不斷更新。希望大家一直關(guān)注中國(guó)腐蝕與防護(hù)網(wǎng)http://www.ecorr.org
責(zé)任編輯:王元
《中國(guó)腐蝕與防護(hù)網(wǎng)電子期刊》征訂啟事
投稿聯(lián)系:編輯部
電話:010-62313558-806
郵箱:fsfhzy666@163.com
中國(guó)腐蝕與防護(hù)網(wǎng)官方 QQ群:140808414
免責(zé)聲明:本網(wǎng)站所轉(zhuǎn)載的文字、圖片與視頻資料版權(quán)歸原創(chuàng)作者所有,如果涉及侵權(quán),請(qǐng)第一時(shí)間聯(lián)系本網(wǎng)刪除。
-
標(biāo)簽: 材料成分, 成分分析, 典型應(yīng)用
相關(guān)文章

官方微信
《中國(guó)腐蝕與防護(hù)網(wǎng)電子期刊》征訂啟事
- 投稿聯(lián)系:編輯部
- 電話:010-62313558-806
- 郵箱:fsfhzy666@163.com
- 中國(guó)腐蝕與防護(hù)網(wǎng)官方QQ群:140808414
文章推薦
點(diǎn)擊排行
PPT新聞

“海洋金屬”——鈦合金在艦船的
點(diǎn)擊數(shù):5768

腐蝕與“海上絲綢之路”
點(diǎn)擊數(shù):4763