取向硅鋼內氧化層形貌對底層物相及形貌的影響
2025-05-29 17:15:52
作者:左暢 來源:腐蝕與防護
分享至:
硅鋼是能量轉換的重要功能材料,硅鋼性能直接 決定了能量轉換效率。取向硅鋼是經過冷軋和退 火工藝后晶粒沿特定方向(通常為軋制方向) 高度一 致排列的硅鋼。取向硅鋼的熱處理工藝流程為脫碳 退火→滲氮→輥涂 MgO→高溫退火→平整拉伸退火 和涂絕緣涂層。其中高溫退火是熱處理時間最長(約7d) ,調控參數最多的一道工序。取向硅鋼表面底層 是在高溫退火過程中由內氧化層與輥涂于表面的 MgO涂層反應生成的具有一定絕緣性的薄膜。通過調控高溫退火參數制備附著性好、厚度均勻的底層是保證取向硅鋼產品獲得優異性能的關鍵。
脫碳退火后取向硅鋼表面輥涂的MgO涂層經烘干后,其內部仍存在部分化合水。這些化合水在高溫退火時會被釋放到鋼卷間隙中,致使層間氧分壓增加,表面脫碳后的內氧化層會繼續氧化,從而影響硅酸鎂底層形貌。研究表明:內氧化層中適量的Fe2SiO4會降低硅酸鎂底層的形成溫度;增加內氧化層中Fe2SiO4含量可提升硅酸鎂底層的連續性,但是過量的Fe2SiO4會使底層易脫落,從而在取向硅鋼表面出現點狀露晶缺陷。
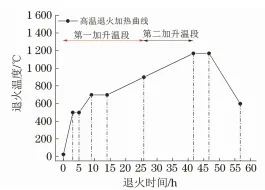
高溫退火過程中取向硅鋼成卷擺放在退火爐內,同一時刻不同區域取向硅鋼卷的溫度是不同的,因此MgO涂層釋放的水蒸氣含量也不相同。依據高溫退火過程中退火氣氛組分變化,高溫退火可分為一次升溫階段(室溫至900℃) 和二次升溫階段 (900~1170℃) 。取向硅鋼表面MgO涂層釋放水蒸氣這一現象發生在一次升溫階段。筆者分別對有無輥涂MgO涂層的取向硅鋼進行高溫退火試驗,探究了露點溫度(Tdp) 對其表面內氧化層及硅酸鎂 底層形貌的影響,并分析了硅酸鎂底層物相及形貌與內氧化層形貌之間的關系,從而確定影響硅酸鎂底層附著性的主要因素。
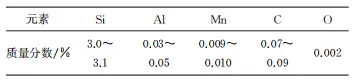
試驗原料為脫碳退火后工業輥涂MgO涂層的取向硅鋼(以下簡稱涂層試樣),以及用酒精將表面MgO輥涂層擦除后的無涂層取向硅鋼(以下簡稱無涂層試樣)。試樣厚度均為0.23mm,經剪板機剪切,制成尺寸為100mm×30mm的試樣,其化學成分如表1所示。
在高溫退火過程中,一次升溫階段退火氣氛中的水氫分壓比(pH2O/pH2) 為0.25,露點溫度分別為5,10,-10℃,二次升溫段N2與H2體積比為1∶3,退火溫度(Ta)分別為900℃和1170℃,高溫退火工藝曲線如圖1所示。高溫退火后,采用 Nova400 Nano型場發射掃描電鏡 (SEM) 觀察試樣表面內氧化層和底層形貌,同時利用8050G型場發射電子探針顯微分析儀(EPMA) 觀察底層的元素組成及分布。
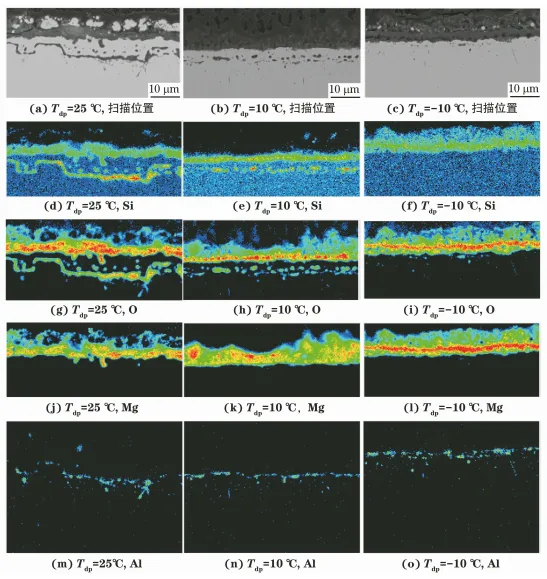
由圖2可見:當退火溫度為900℃時,涂層試樣表面尚未形成底層,隨著露點溫度由25℃降至-10℃,退火氣氛中的氧分壓逐漸降低,此時內氧化層厚度由8.81μm降低至2.86μm;當退火溫度為1170℃,露點溫度為25℃時,涂層試樣表面形成了附著性良好的底層,且在底層下方形成了連續的帶狀氧化物,在帶狀氧化物與底層間形成了平均直徑約1.49μm的球形氧化物顆粒;當露點溫度為10℃時,涂層試樣表面形成了附著性較好的底層,且在底層下方形成了平均直徑約0.59μm的球形氧化物顆粒;當露點溫度為-10℃時,涂層試樣表面形成的底層與基體發生剝離,底層下方形成了平均直徑約0.23μm的球形氧化物顆粒。
圖 2 不同露點溫度下涂層試樣經高溫退火后的截面內氧化層及底層的微觀形貌
由圖3可見:在1170℃退火溫度下,當露點溫度分別為25,10,-10℃時,涂層試樣表面底層均由Mg、Si及O元素組成,氧化物主要為Mg2SiO4;當露點溫度為25℃時,底層下方球形氧化物主要由Si和O元素組成,氧化物為SiO2,其下方連續的帶狀氧化物主要由Si、O及Al元素組成,氧化物為Al2O3- SiO2系莫來石;當露點溫度為10℃時,底層下方球形氧化物顆粒主要為Al2O3-SiO2系莫來石;當露點溫度為-10℃時,底層下方球形氧化物顆粒非常小,此時在底層與基體的界面處也形成了Al2O3- SiO2系莫來石。
圖 3 不同露點溫度下涂層試樣經 1170℃退火后底層截面的元素掃描位置和掃描結果
由圖4可見:當退火溫度為900℃時,隨著露點溫度從25℃降低至-10℃,退火氣氛中的氧分壓逐漸降低,無涂層試樣表面內氧化層厚度由8.05μm降低至3.49μm;當退火溫度為1170℃時,隨著露點溫度由25℃降低至-10℃,內氧化層厚度由12.81μm降低至3.32μm,球形氧化物平均顆粒尺寸由0.92μm減小到0.25μm,且在球形氧化物顆粒、硅鋼與退火氣氛界面處均存在鐵層。
圖4 不同露點溫度下無涂層試樣經高溫退火后內氧化層的截面微觀形貌
2. 3. 1 內氧化層演化規律
根據Wagner氧化理論, 內氧化層中氧化物形 貌主要受氧化前沿遷移速率v控制。氧化前沿遷移速率用公式(1)表示。
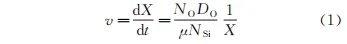
式中:t為氧化時間;NO是固溶在取向硅鋼與退火氣氛界面上氧的摩爾分數;DO是氧元素在硅鋼中的擴散系數;NSi是硅鋼基體中硅的摩爾分數;X是氧化前沿距硅鋼與退火氣氛界面的距離;μ為與材料相關的常數,μ取值為2.
對于無涂層取向硅鋼,其氧化物形貌變化的主要影響因素是固溶進入鋼中的氧含量,其主要來自退火氣氛中的水蒸氣;對于有涂層取向硅鋼,影響其氧化物形貌的因素除了氣氛中的水蒸氣,還有來自于涂層中的氧。當取向硅鋼表面無MgO涂層時,在表面內層氧化過程中會先形成球狀氧化物,之后隨內氧化層厚度的增加,氧化前沿遷移速率也逐漸降低,此后才會逐漸形成片層狀氧化物并連成帶狀氧化物。當取向硅鋼表面涂覆MgO涂層時,退火氣氛中水蒸氣需先穿過MgO涂層才能到達取向硅鋼表面,與無涂層取向硅鋼相比,有涂層取向硅鋼表面內氧化層中氧化物形貌主要為片層狀和帶狀,而不再是球狀和帶狀。這說明在本試驗條件下與無涂層試樣相比,涂層試樣表面內氧化層的氧化前沿遷移速率更低。根據式(1) 可見,在相同退火溫度與氧化深度條件下,取向硅鋼的DO、NSi及X均是定值,此時v與NO呈正比。 因此,MgO涂層的存在會阻礙水蒸氣在取向硅鋼表面固溶,使其表面固溶氧含量下降。
2.3.2 內氧化層形貌對底層物相及形貌演化的影響
對于無涂層試樣,當退火溫度高于900℃時,退火氣氛組成為25%(體積分數)N2+75%(體積分數) H2,此時退火氣氛中的氧分壓逐漸趨于零。從Si-O系蒸發物質圖上可以看出,當氣氛中的氧分壓接近于SiO2的分解壓力時,與SiO2和Si相平衡的SiO的揮發活性逐漸增強。在此條件下,無涂層試樣表面SiO2逐步分解為SiO并揮發,當退火溫度為1170℃時,表層轉變為鐵層。當露點溫度為25℃時,無涂層試樣表面內氧化層最厚;隨著退火溫度的升高,只有表層SiO2分解揮發,當退火溫度達到1170℃時,基體內部仍嵌有未分解的大顆粒氧化物 [圖5(a)]。當露點溫度為-10℃時,試樣表面內氧化層最薄,在退火溫度高于900℃時,試樣表層大部分氧化物顆粒均分解揮發,當退火溫度達到1170℃時,基體內只留有部分尺寸非常小的氧化物顆粒。
對于涂層試樣,涂層將退火氣氛與內氧化層隔離,這在一定程度上抑制了SiO2分解揮發。對比圖2(a,c, e) 與圖2(b, d, f) 可知,最終生成的底層厚度比內氧化層薄。因此,在底層形成過程中,除了Mg2+向基體內擴散,SiO2也會向試樣表面遷移。當露點溫度為25℃時,涂層試樣表面內氧化層最 厚,由EPMA圖譜可知,此時Mg2+并未擴散至內氧化層與基體的界面,底層下方仍有球狀及連續的帶狀SiO2,此后它們會與基體中抑制劑AlN分解出的Al反 應 生成 Al2O3-SiO2 系 莫 來 石 [ 圖 5 (b)] 。當露點溫度為-10℃時,涂層試樣表面內氧化層最薄,此時大部分氧化物顆粒與遷移至內氧化層的Mg2+發生反應,因此在底層下方生成的球形莫來石顆粒尺寸非常小。上述試驗結果表明,在高溫退火過程中涂層取向硅鋼表面底層下方氧化物形貌的演變規律與無涂層取向硅鋼表面內氧化層與基體界面的氧化物形貌一致。影響取向硅鋼表面氧化物形貌演化的主要因素是內氧化層厚度。
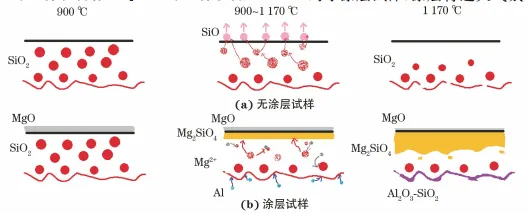
圖 5 有無涂層試樣表面內氧化層及底層形貌演化示意
在底層形成過程中,當底層下方形成較大尺寸的球形或連續的帶狀氧化物時,Al在向取向硅鋼表面遷移時會先擴散至底層與基體界面并與氧化物反應,這會改變底層與基體界面莫來石的分布,而物相組成的不同會造成底層與基體存在熱膨脹差異。硅 酸鎂的熱膨脹系數為 11×10-6K-1,莫來石的熱膨脹系數為5.3×10-6K-1,基體的熱膨脹系數為15×10-6K-1,底層中的莫來石數量越多,底層與基體間熱膨脹系數的躍變越大。這會促使底層產生更大的熱應力,導致底層更易發生開裂和剝離。因此,當底層下方球形氧化物顆粒數量及尺寸非常小時[圖 2 (f)],莫來石易聚集在底層與基體的界面,此時底層易與基體剝離。
(1) 當高溫退火溫度為900℃ ,水氫分壓比為0.25,露點溫度由25℃降至-10℃時,無涂層試樣表面內氧化層中氧化物顆粒均呈球狀與帶狀,內氧化層厚度由8.05μm 降低至3.49μm。
(2) 當退火溫度為900℃,露點溫度由25℃降至-10℃時,涂層試樣表面內氧化層厚度由8.81μm減薄至2.86μm,此時試樣表面沒有形成底層。當退火溫度達到1170℃時,試樣表面形成了底層,隨著露點溫度的降低,底層下方球形氧化物顆粒直徑由1.49μm減小至0.23μm。當底層下方界面的Al2O3-SiO2系莫來石分布會減少,因熱膨脹系數不匹配造成的底層剝落問題也會相應減少。
(3) 當退火溫度達到1170℃時,涂層試樣表面底層下方球形氧化物形貌的演變規律與無涂層試樣表面內氧化層與基體界面的氧化物形貌一致。影響其形貌演化的主要因素是底層形成前內氧化層的厚度。
免責聲明:本網站所轉載的文字、圖片與視頻資料版權歸原創作者所有,如果涉及侵權,請第一時間聯系本網刪除。